UNA METODOLOGÍA PARA LA CARACTERIZACIÓN MOLECULAR DE LOS VIRUS ENDÉMICOS, EMERGENTES Y DE POTENCIAL EMERGENCIA EN LA REGIÓN DE COCLÉ DEL NORTE
Chang, Jim
Universidad Tecnológica de Panamá
Ciudad de Panamá, Panamá
jim.chang@utp.ac.pa
Sánchez-Galán, Javier E.
Universidad Tecnológica de Panamá
Ciudad de Panamá, Panamá
javier.sanchezgalan@utp.ac.pa
Martínez, Alexander
Instituto Conmemorativo Gorgas en Estudios de la Salud
Ciudad de Panamá, Panamá
almartinez@gorgas.gob.pa
Díaz, Yamilka
Instituto Conmemorativo Gorgas en Estudios de la Salud
Ciudad de Panamá, Panamá
ydiaz@gorgas.gob.pa
Wallau, Gabriel
Fundação oswaldo cruz
Pernambuco, Brasil
gabriel.wallau@fiocruz.br
https://doi.org/10.33412/apanac.2025.82
Abstract
Panama acts as a biological bridge for the American continent, which gives it a high diversity of pathogens due to its geographical position, tropical climate, transit of migratory species, and high human mobility. Viruses such as dengue, Zika, and chikungunya, along with others with emergency potential, have a significant impact on the morbidity and mortality of the population, in addition to representing an economic and social burden on the health system.
The Coclé del Norte region has become a critical point in this context due to mining activity in the area. These activities have affected biodiversity and natural resources, altering environmental conditions and forcing the displacement of endemic species to other regions, facilitating the spread of these pathogens. This project aims to characterize the viral diversity present in reservoirs using state-of-the-art techniques in sequencing, metagenomics, and bioinformatic analysis.
Biological samples were collected from wild reservoirs, mainly bats. For the identification and characterization of viruses present, massive sequencing techniques and bioinformatics tools focused on metagenomics were applied. The analysis of the data generated provided preliminary results with information on the microbial composition of the reservoirs, as well as relevant data to advance the discovery of potentially dangerous viruses.
Keywords: Metagenomics, Virome, Reservoirs, Biodiversity, Pathogens.
Resumen
Panamá funciona como puente biológico del continente americano, lo que le confiere una alta diversidad de patógenos debido a su posición geográfica, clima tropical, tránsito de especies migratorias y alta movilidad humana. Virus como el dengue, zika y chikungunya, junto con otros de potencial emergencia, tienen un impacto significativo en la morbilidad y mortalidad de la población, además de representar una carga económica y social para el sistema de salud.
La región de Coclé del Norte se ha convertido en un punto crítico en este contexto debido a la intervención minera en la zona. Estas actividades han afectado la biodiversidad y los recursos naturales, alterando las condiciones ambientales y forzando el desplazamiento de especies endémicas hacia otras regiones, facilitando la propagación de estos patógenos. Este proyecto tiene como objetivo caracterizar la diversidad viral presente en reservorios mediante las técnicas de última generación en secuenciación, metagenómica y análisis bioinformático.
Se recolectaron muestras biológicas de reservorios silvestres, principalmente de murciélagos. Para la identificación y caracterización de virus presentes, se aplicaron técnicas de secuenciación masiva y herramientas bioinformáticas enfocadas en metagenómica. El análisis de los datos generados permitió obtener resultados preliminares con información sobre la composición microbiana de los reservorios, así como datos relevantes para avanzar en el descubrimiento de virus potencialmente nuevos, su posible capacidad patogénica y sus patrones de evolución viral.
Este estudio permitirá generar un conocimiento integral sobre la diversidad viral en reservorios silvestres de la región de Coclé del Norte, contribuyendo a la identificación temprana de virus emergentes y potencialmente patógenos.
Palabras claves: Metagenómica, Viroma, Reservorios, Biodiversidad, Patógenos.
1. Introducción
Nuestro país constituye una alta complejidad ecológica, caracterizado por una convergencia de ecosistemas tropicales que favorecen la diversidad de microorganismos, como los virus de importancia epidemiológica [1]. Su posición como corredor biológico, sumada a patrones climáticos tropicales, movilidad humana intensa y tránsito de especies migratorias, genera un escenario propicio para la introducción, mantenimiento y circulación de patógenos emergentes y reemergentes [2]. Estos factores en conjunto, convierten a Panamá en un punto crítico para el estudio de dinámicas virales y procesos de transmisión [3].
Panamá ha desempeñado un rol central en la investigación de arbovirus, aportando evidencia esencial para el desarrollo de estrategias de control vectorial durante el siglo XX [4]. Sin embargo, la reintroducción sostenida de dengue desde la década de 1990 y la incursión de chikungunya y Zika han modificado significativamente la ecología viral regional, evidenciando la limitada capacidad del sistema de vigilancia tradicional para caracterizar escenarios de co-circulación y sintomatología superpuesta. Esta debilidad metodológica se reflejó en la epidemia de Zika de 2015–2017, donde la ausencia de un enfoque genómico integrado dificultó la discriminación entre patógenos coexistentes, generando subdiagnóstico y errores en la vigilancia epidemiológica [5],[6].
La metagenómica se ha consolidado como una herramienta clave para la detección no dirigida de virus, permitiendo explorar el viroma de diversas matrices biológicas, identificar agentes infecciosos conocidos y novedosos, y estimar patrones evolutivos con alta resolución [7],[8]. Este estudio propone un enfoque interdisciplinario que combina secuenciación masiva, análisis bioinformático y modelos matemático-epidemiológicos para caracterizar la filogeografía y la co-circulación viral en Panamá, fortaleciendo la capacidad nacional de vigilancia y respuesta ante virus endémicos, emergentes y de potencial emergencia.
2. método
1. RECOLECCIÓN Y PROCESAMIENTO DE MUESTRAS
Las muestras biológicas fueron obtenidas a partir de murciélagos capturados en sitios definidos según criterios ecológicos y gradientes de perturbación. Se aplicaron protocolos estandarizados para garantizar bioseguridad y calidad del material. La extracción de ácidos nucleicos se realizó mediante kits comerciales, incorporando controles internos y cuantificación, asegurando el rendimiento e integridad del material genético.
2. SECUENCIACIÓN Y ANÁLISIS METAGENÓMICO
Las librerías fueron preparadas siguiendo el protocolo optimizado “Unbiased metagenomic protocol: from RNA to sequencing” [9] del Departamento de Genómica y Proteómica del ICGES para plataformas de tercera generación. Las lecturas crudas fueron evaluadas con FastQC, filtradas y recortadas mediante fastp, normalizadas en longitud con chopper y formateadas usando reformat. El ensamblaje se realizó con SPAdes, seguido de la clasificación taxonómica con Kraken2 y la confirmación de identidad mediante BLAST.
3. Resultados
Los resultados de este análisis preliminar se muestran en las Figuras 1 y 2.
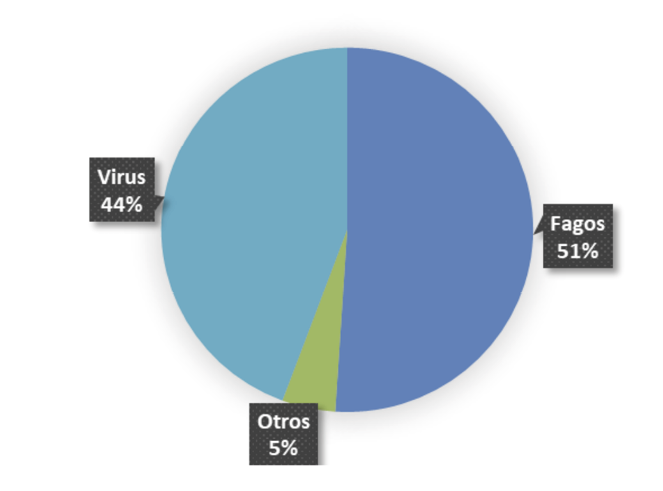
Fig. 1. Composición taxonómica global del metagenoma (porcentaje de lecturas clasificadas)
Proporción porcentual de lecturas clasificadas en muestras de reservorios (murciélagos), determinada mediante clasificación taxonómica con Kraken2. Los resultados muestran predominio de fagos (51%), seguido por virus no bacterianos (44%) y otros (5%), indicando una alta representación bacteriana en las muestras y una fracción viral relevante para análisis ecológicos y zoonóticos.
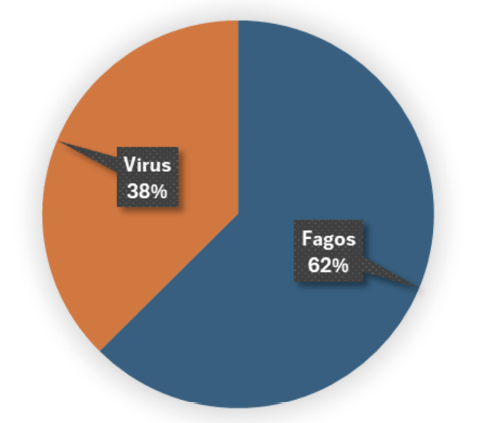
Fig. 2. Distribución de secuencias largas y de alta similitud por categoría
Porcentaje de secuencias más largas y con mayores índices de similitud taxonómica, analizadas tras ensamblaje y clasificación. El 62% corresponde a fagos y el 38% a virus con homología reportada en mamíferos, lo que sugiere la presencia de secuencias virales de interés zoonótico en las muestras.
4. CONCLUSIONES
Los análisis metagenómicos revelan un predominio de fagos en las muestras, tanto en la distribución global de lecturas (51%) como en las secuencias más largas y con mayor similitud (62%). Este patrón sugiere una elevada actividad bacteriana en los reservorios y resalta el papel ecológico de los fagos como moduladores claves de las comunidades microbianas. La detección de virus no bacterianos que constituyen el 44% de las lecturas clasificadas y el 38% de las secuencias de mayor homología indica la presencia de una fracción viral con posible relevancia zoonótica.
La identificación de secuencias con similitud a virus reportados en mamíferos no solo refleja la sensibilidad del enfoque metagenómico, sino que también sugiere, de manera plausible y consistente, que los murciélagos podrían actuar como reservorios de virus de ARN.
Hacia el futuro se prevé que la detección de estas secuencias refleje la presencia de virus reales o de variantes altamente similares de relevancia para la vigilancia viral nacional. Se avanzará en el análisis de las secuencias no clasificadas para permitir explorar la existencia de virus aún no descritos, así como desarrollar y evaluar modelos matemáticos o computacionales para estudiar la dinámica de circulación y co-circulación viral.
Referencias
- N. Myers, R. A. Mittermeier, C. G. Mittermeier, G. A. B. da Fonseca, and J. Kent, ‘Biodiversity hotspots for conservation priorities’, Nature, vol. 403, no. 6772, pp. 853–858, Feb. 2000, doi: 10.1038/35002501.
- M. K. Parvez and S. Parveen, ‘Evolution and Emergence of Pathogenic Viruses: Past, Present, and Future’, Oct. 01, 2017, S. Karger AG. doi: 10.1159/000478729.
- D. Franco et al., ‘Early Transmission Dynamics, Spread, and Genomic Characterization of SARS-CoV-2 in Panama’, Emerg Infect Dis, vol. 27, no. 2, pp. 612–615, Feb. 2021, doi: 10.3201/eid2702.203767.
- Y. Díaz et al., ‘Molecular epidemiology of dengue in Panama: 25 years of circulation’, Viruses, vol. 11, no. 8, Aug. 2019, doi: 10.3390/v11080764.
- P. C. S. Farias et al., ‘Epidemiological profile of arboviruses in two different scenarios: dengue circulation vs. dengue, chikungunya and Zika co-circulation’, BMC Infect Dis, vol. 23, no. 1, Dec. 2023, doi: 10.1186/s12879-023-08139-6.
- R. J. Oidtman, G. España, and T. Alex Perkins, ‘Co-circulation and misdiagnosis led to underestimation of the 2015–2017 zika epidemic in the americas’, PLoS Negl Trop Dis, vol. 15, no. 3, Mar. 2021, doi: 10.1371/journal.pntd.0009208.
- J. V. C. Souza et al., ‘Viral Metagenomics for the Identification of Emerging Infections in Clinical Samples with Inconclusive Dengue, Zika, and Chikungunya Viral Amplification’, Viruses, vol. 14, no. 9, Sep. 2022, doi: 10.3390/v14091933.
- H. Cholleti, M. Berg, J. Hayer, and A. L. Blomström, ‘Vector-borne viruses and their detection by viral metagenomics’, Jan. 01, 2018, Taylor and Francis Ltd. doi: 10.1080/20008686.2018.1553465.
- A. Martínez, C. González, S. Whitmer, O. Chavarría, J. Góndola, and A. Moreno, ‘Unbiased metagenomic protocol: from RNA to sequencing’, Panamá.
Agradecimientos
Agradezco a la Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) por el apoyo brindado a través del Programa de Doctorado en Biociencias y Biotecnología, en el cual soy estudiante becario. Asimismo, expreso mi agradecimiento a mis tutores, Javier Sánchez Galán y Alexander Martínez, por su acompañamiento académico y por el respaldo financiero proporcionado mediante sus fondos del Sistema Nacional de Investigación (SNI), que han sido fundamentales para el desarrollo de este trabajo.
Autorización y Licencia CC
Los autores autorizan a APANAC XVIII a publicar el artículo en las actas de la conferencia en Acceso Abierto (Open Access) en diversos formatos digitales (PDF, HTML, EPUB) e integrarlos en diversas plataformas online como repositorios y bases de datos bajo la licencia CC:
Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) https://creativecommons.org/licenses/by-nc-sa/4.0/.
Ni APANAC XVIII ni los editores son responsables ni del contenido ni de las implicaciones de lo expresado en el artículo.